So ein Pech aber auch! Da unterlaufen führenden Wissenschaftlern der Alzheimer-Forschung ständig dumme Flüchtigkeitsfehler, die ihre Forschung wertlos machen, während sie durch ihre einflussreichen Positionen die Zukunft dieses Forschungsfeldes bestimmen.
Haben Sie zufällig ein paar Euro auf der hohen Kante und möchten diese gewinnbringend in ein Unternehmen investieren? Dann habe ich hier was für Sie. Die Ramaswamy-Methode. Und sie ist kinderleicht:
Zunächst suchen Sie sich ein Unternehmen, das vergeblich versuchte, ein Medikament gegen Alzheimer zu entwickeln. Geben wir dem Mittel den Namen Intepirdine. Je öfter dieses Medikament in klinischen Studien gescheitert ist, desto besser. So hat GlaxoSmithKline (GSK) bereits 4x erfolglos versucht, den Nutzen des Mittels nachzuweisen. Erwerben Sie nun die Rechte an diesem Wirkstoff für 5 Millionen Dollar. In der Pharmaindustrie sind dies Centbeträge.
Nun gründen Sie ein Pharmaunternehmen – nennen wir es Axovant -, mit dem Sie neue klinische Studien anmelden, durch die Sie die Wirksamkeit von Interpirdine nachweisen wollen. Zum Glück ist ihre eigene Mutter Geriaterin, sodass Sie nun eine Person haben, die keinerlei persönliche Verbindung zu Ihnen hat und gleichzeitig bei der Durchführung der klinischen Studien als Beraterin zur Seite stehen kann. Denn nur weil ein Mittel bereits 4x gescheitert ist, heißt das nicht, dass das 5. Mal nicht den Durchbruch bringt. Und jetzt folgt der wichtigste Schritt:
Noch bevor die klinischen Studien abgeschlossen sind, erzeugen Sie einen riesigen Hype um das Medikament, sodass Investoren glauben müssen, Sie haben einen Erfolg in der Hand, der nur noch realisiert werden muss. Mit diesem Hype im Rücken bringen Sie das Unternehmen in einem der erfolgreichsten Börsenstarts der Biotechnologiebranche an die Öffentlichkeit. Selbstverständlich kaufen Sie über Ihre Muttergesellschaft, Roivant Sciences, ordentlich Anteile, die zeitnah – und natürlich gewinnbringend – verkauft werden, noch bevor Daten zur Wirksamkeit des Medikaments vorliegen. Wenn das Medikament dann plötzlich und überraschend scheitert, die Aktie 75% ihres Wertes über Nacht verliert, was Ihr Unternehmen quasi wertlos macht, sind Sie und Ihre Investitionen durch dieses Konstrukt trotzdem geschützt.
Beim Finanzamt knallen nun die Korken, weil Sie für dieses Geschäftsjahr Einnahmen in Höhe von 37 Millionen US-Dollar anmelden können. Herzlichen Glückwunsch! Sie sind über Nacht zum Millionär geworden (vor Steuern). Und das vollkommen legal!
Benannt habe ich diese Taktik nach Vivek Ramaswamy, dem ehemaligen Pharmaunternehmer, republikanischen Präsidentschaftskandidaten und zukünftigem Leiter der „Kommission zur Deregulierung und Senkung der Staatsausgaben“ unter Präsident Trump. Zumindest sollte er das werden, denn noch in der (notorisch viel zu langen) Entstehungsphase dieses Beitrags wurde er wieder dem Arbeitsmarkt zugeführt. Natürlich hat Ramaswamy mit seinem Unternehmen nur versucht, seinen Teil zur Heilung von Alzheimer beizutragen, daher kann man ihm selbstverständlich in keinem der Punkte, in denen ich ihm hier einen Vorwurf mache, einen Vorwurf machen. In der Pressemeldung des Unternehmens steht immerhin auch, dass man enttäuscht und traurig sei, dass sich die Hoffnungen auf eine Wirksamkeit des Medikaments zerschlagen haben. Und am eigentlichen Medikament kann man – und das meine ich ausnahmsweise ganz unironisch – nichts aussetzen. Klar, es wirkt nicht, aber das ist der Weg, den Wirkstoffe gehen. Wenn es sich in klinischen Tests nicht beweist, wird die Weiterentwicklung eingestellt. Anschließend steht es jedem Käufer frei, die Rechte am Wirkstoff zu erwerben und eigene Untersuchungen durchzuführen. Nochmal: Das ist ein herkömmlicher Weg, den man in der Forschung geht. Man kann sich entscheiden, Holzwege zu betreten und sie konsequent bis ans Ende zu gehen. Denn manchmal entpuppt sich der vermeintliche Holzweg eben doch als Milliarden-Dollar-Idee. Und manchmal entpuppt sich der Holzweg nicht nur als Holzweg, sondern als schnellster Weg in den Knast.
Wie man 500 Millionen Euro verbrennt
Den Erfolg eines Medikaments vorherzusehen ist beinahe unmöglich. Einen Misserfolg zu wittern ist hingegen manchmal einfacher. Und in diese Kategorie fällt Simufilam, das Medikament, das von Cassava Sciences entwickelt wurde. Als die Behörden die Zulassung wie erwartet verwehrten, riss dies den Aktienkurs innerhalb kürzester Zeit von $15,50 auf $2,50 und damit den Unternehmenswert quasi über Nacht von rund $620 Millionen auf $100 Millionen Dollar.
Warum diese Kursentwicklung für Fachleute nicht überraschend kam, hat viel damit zu tun, was wir über Alzheimer wissen – und was nicht. Aber noch wichtiger sind die Rollen von Hoau-Yan Wang, und Lindsay Burns, den Wissenschaftlern, deren Forschung Grundlage von Simufilam ist.
Gegen Dr. Wang ermittelt mittlerweile das FBI, da er verdächtigt wird, seine Forschungsergebnisse manipuliert und so über $16 Millionen Fördermittel erschlichen zu haben.
Ein Vorwurf, der gegen Dr. Burns erhoben wird ist, dass sie Forschungsergebnisse einer Phase-2-Studie zu Simufilam manipuliert haben soll, um dem Medikament eine Wirksamkeit zu attestieren, die dieses nicht haben konnte. Aber immer der Reihe nach:
Ziel von Simufilam war das Protein Filamin A. Dieses Protein spielt eine wichtige Rolle bei der strukturellen Integrität des Zellaufbaus, indem es Aktinfilamente im Zytoplasma vernetzt. Aktin gibt der Zelle ihre Struktur, die Vernetzung durch Filamin A verbindet die Aktinfilamente und stabilisiert sie. Die Idee war, dass Filamin A in Alzheimerpatienten entartet ist. Dieses Filamin A sollte – so die Idee – an den nikotinischen Rezeptor α7nAChR binden, dessen Signalübermittlung bei Alzheimerpatienten gestört ist. Wirken sollte Simufilam, indem es das Filamin A in eine normale Konformation zurückführt, um die gestörten Signalwege zu normalisieren. Cassava Sciences konnte allerdings nie nachweisen, dass diese Arbeitshypothese korrekt ist. Es kommt aber noch besser, denn inzwischen konnten mehrere Arbeiten zeigen, dass nikotinische Rezeptoren sich nicht zur Alzheimertherapie eignen, weshalb dieses Target nicht mehr weiterverfolgt wird.
Darüber hinaus konnte das Unternehmen keine Daten vorzeigen, die bestätigen, dass Simufilam überhaupt an Filamin A bindet. Allerdings wäre dieser Part der Entscheidende gewesen, um Vertrauen in das Medikament zu wecken. Bei der Interaktion zwischen Filamin A und Aktin handelt es sich um eine Protein-Protein-Wechselwirkung. Proteine sind große, komplexe Moleküle, die aus diversen Untereinheiten bestehen. Diese Komplexität ist nötig, da Proteine häufig nur einen Zweck haben – nämlich ein ganz bestimmtes Molekül zu binden und zu verarbeiten. Damit auch nur dieses Molekül binden kann, muss die Affinität zwischen Molekül und Protein extrem hoch sein. Für Protein-Protein-Interaktionen gilt dies umso mehr. Denn hier muss nicht nur ein Molekül an ein Protein binden, sondern zwei Proteine aneinander. Wer nun einen Wirkstoff designen will, der diese intensiven Wechselwirkungen unterbricht, muss nicht nur darauf achten, dass sein Wirkstoff an das Protein bindet, er muss auch zeigen, dass diese Bindung so stark ist, dass das andere Protein nicht mehr binden kann.
Wer nicht zeigen kann, dass sein Wirkstoff überhaupt eine Wirkung hat, der kann auch nicht untersuchen, mit welchen chemischen Modifikationen des Wirkstoffs diese Wirkung noch gesteigert werden kann.
Aber vielleicht war eine zu genaue Untersuchung des Wirkstoffes ja auch nie gewollt. Es ist Zeit, dass wir uns intensiver mit Wang und Burns beschäftigen.
Wang und Burns hatten bereits vor ihrer Zeit bei Cassava Sciences eine gemeinsame Geschichte bei Pain Therapeutics, dem Unternehmen, das 1998 von Cassavas zukünftigem CEO (und späterem Ehemann von Lindsay Burns) gegründet wurde. Hier untersuchten Wang und Burns die Idee, ob man die Abhängigkeit von opioidhaltigen Schmerzmitteln reduzieren und gleichzeitig die Schmerzlinderung steigern kann, wenn man das Opioid gemeinsam mit einem Opioid-Blocker verabreicht. Sie wollten damit das Missbrauchspotenzial verringern, in dem der Blocker aktiv wird, wenn das Opioid auf anderem Wege (z.B. per Injektion) verabreicht wird.
Um sein gescheitertes Projekt zu retten, versuchte Wang eine Erklärung zu finden, wieso seine Idee trotzdem funktionieren konnte. Hier kam zum ersten Mal Filamin A ins Spiel, das angeblich mit dem Opioid-Blocker wechselwirken sollte. Sein 2008 veröffentlichtes Paper, in dem dieser Effekt nachgewiesen werden sollte, wurde mittlerweile wegen „Unregelmäßigkeiten“ zurückgezogen.
Allerdings sollten dies nicht die einzigen Unregelmäßigkeiten in seiner Arbeit bleiben. Da wir im Laufe des Artikels noch einige weitere Wissenschaftler und die „Unregelmäßigkeiten“ ihrer Arbeit betrachten wollen, raffen wir an dieser Stelle die Ereignisse ein wenig:
Filamin A wurde zu einem beliebten Studienobjekt von Wang. Als es allerdings darum ging, ihr Alzheimermedikament Simufilam zuzulassen, erkannten er und die verantwortlichen Wissenschaftler, dass ihr Alzheimermedikament nur in geringer Dosis im Gehirn gefunden werden konnte. Das Medikament, dessen hypothetischer Wirkmechanismus nie nachgewiesen wurde, wird nur in geringer Dosis dort gefunden, wo es überhaupt wirken soll. Aber es kommt noch schlimmer.
Dr. Burns wird vorgeworfen, Daten von 40% der Studienteilnehmer aus einer Phase-2-Studie entfernt zu haben, nachdem sie Kenntnis erlangt hatte, welche Teilnehmer das Medikament und welche das Placebo erhalten haben. Durch diese Handlung schien das Medikament in der entsprechenden Studie wirksamer als das Placebo.
Des Weiteren wurden 5 Studien von Cassava Sciences, an denen Wang beteiligt war, zurückgezogen. Außerdem wird gegen Wang ermittelt, weil er nicht in der Lage ist, die Rohdaten seiner Studien vorzulegen. Auf der Website cassavafraud.com wird in mehreren Dokumenten ausführlich auf die Vorwürfe eingegangen.
Aber selbst, wenn wir es nicht mit Wissenschaftlern und den „Unregelmäßigkeiten“ in ihrer Arbeit zu tun haben, ist die Erforschung von Alzheimer und die Entwicklung eines Medikaments eine herausfordernde Aufgabe.
Warum es so schwierig ist, Alzheimer zu therapieren
Wir wissen nicht, wie Alzheimer entsteht. Wüssten wir es, gäbe es zumindest einen klaren Erkrankungspfad (sprich: Pathomechanismus), der uns Möglichkeiten zur Entwicklung neuer Medikamente bietet. Und obwohl wir diesen Pathomechanismus nicht kennen, gibt es eine Arbeitshypothese, die zumindest teilweise korrekt sein muss. Ich habe die Hoffnung, dass ich innerhalb der nächsten Jahre den folgenden Abschnitt anpassen muss, weil wir den Mechanismus aufgeklärt haben, aber bis dahin ist das die Essenz dessen, was wir über Alzheimer wissen:
Das Amyloid-Vorläuferprotein (APP) ist ein Transmembranprotein, also ein Protein, das die Zellmembran durchdringt, um das Innere und Äußere der Zelle zu verbinden. Wir finden es in vielen Geweben des Körpers, besonders im zentralen Nervensystem. Seine biologische Funktion ist noch nicht vollständig verstanden, aber es gibt eine Reihe von Hinweisen. So wird das Protein mit der Bildung, Vernetzung und Regulation von Synapsen in Verbindung gebracht. Der intrazelluläre (innere) Bereich von APP interagiert mit Proteinen, die am Transport von Molekülen innerhalb der Zelle beteiligt sind, insbesondere entlang der Mikrotubuli. Dies ist wichtig für die Versorgung von Neuronen mit Nährstoffen und anderen Substanzen. Aber dazu später mehr.
Ein Protein besteht aus einer langen, in sich verwundenen Kette von Aminosäuren. Diese Kette kann an einer beliebigen Stelle zerschnitten werden, um so neue Proteine oder Peptide (kurze Proteine) zu erzeugen. Wenn Proteine wie APP das Ende ihrer Lebensdauer erreicht haben, geschieht dieser Prozess z.B. um die Aminosäuren zu recyclen. Im Falle von APP sind die Aufgaben seiner gespaltenen Proteine aber noch zahlreicher.
APP wird von den Enzymen α-Sekretase und γ-Sekretase in die Fragmente sAPPα und AICD gespalten. sAPPα dient dem Zellwachstum und dem Schutz vor oxidativem Stress, AICD beeinflusst die Genexpression im Inneren der Zelle.
Bis zu diesem Punkt funktioniert der biochemische Prozess exakt so, wie er funktionieren soll. Das Protein wird korrekt geschnitten, die Fragmente funktionieren, alles tutti.
Allerdings wird APP ebenfalls vom Enzym β-Sekretase anstelle der α-Sekretase gespalten. Es entstehen nun neue Fragmente aus unserem APP, von denen besonders Amyloid-β 42 (Aβ42) wichtig ist. Dieses Peptid sammelt sich im Laufe der Zeit in Form der Amyloid-Plaques an, von denen im Zusammenhang mit Alzheimer oft die Rede ist. Diese Plaques stimulieren entzündliche Prozesse im Gehirn und beeinträchtigen die Funktion der Synapsen. Genau aus diesem Grund existieren verschiedene Enzyme im Körper, die diese Plaques abbauen, um den Entzündungen und damit verbundenen Schädigungen entgegenzuwirken. Eigentlich wäre dieser Mechanismus eine wunderbare Erklärung für die Entstehung von Alzheimer. Leider gibt es da zwei große Probleme:
Es gibt Menschen, die diese Plaques in hoher Menge im Gehirn haben und keine Symptome von Alzheimer zeigen. Und Medikamente, die diese Plaques entfernen sollen, zeigen ebenfalls nur sehr, sehr schwachen, klinischen Erfolg bei der Linderung oder Vermeidung von Alzheimer. Der Erfolg ist so gering, dass sich kaum sagen lässt, ob er wirklich existiert.
Daher musste die Amyloid-Hypothese erweitert werden, da sie offensichtlich als Erklärung unzureichend ist.
Tau-Hypothese
Unsere Zellen sind von sogenannten Mikrotubuli durchzogen. Diese stabilisieren die Zelle und dienen gleichzeitig als Transportsystem für z.B. Nährstoffe oder Neurotransmitter. Da eine Zelle kein starres Konstrukt ist, sondern bis zu einem gewissen Grad verformt werden kann (z.B. bei der Bildung neuronaler Verknüpfungen beim Lernen), müssen die Mikrotubuli ebenfalls flexibel sein. Um dies zu gewährleisten, existiert das Tau-Protein. In seinem „normalen“ Zustand bindet es eng an die Mikrotubuli und gibt ihnen so ihre Stabilität.
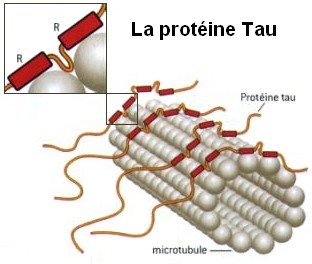
Wenn eine gewisse Flexibilität der Zelle benötigt wird, muss das Tau-Protein die Mikrotubuli „loslassen“. Dies geschieht, indem Kinasen (eine Gruppe von Enzymen) eine Phosphatgruppe an das Protein anbringen. Dies reduziert die Affinität des Tau-Proteins zu den Mikrotubuli, wodurch es nicht mehr an diese bindet. Dieser Prozess, die Phosphorylierung, wird von einer weiteren Enzymgruppe, den Phosphatasen gesteuert, die bei Bedarf diese Phosphatgruppe wieder vom Tau-Protein entfernen und so für eine bessere Bindung an die Mikrotubuli sorgen können.
Die derzeitige Hypothese, wie dieser Mechanismus zu Alzheimer führen kann, ist folgende:
Durch die Amyloid-Plaques werden chronische Entzündungsprozesse ausgelöst, die das spezielle Immunsystem des Gehirns auf den Plan rufen. Genauer: Die sogenannten Mikroglia. Zur Aktivierung der Immunzellen (u.a. der Mikroglia) wird das Enzym GSK-3β benötigt, das gleichzeitig auch das Tau-Protein phosphoryliert. Je mehr Mikroglia aktiv sein müssen, desto mehr GSK-3β ist in den Zellen vorhanden, was die Tau-Proteine immer stärker phosphoryliert und so ihre Affinität für Mikrotubuli reduziert. Je weniger Tau-Proteine an die Mikrotubuli binden, desto geringer ist ihre strukturelle Integrität. Wird ein Schwellenwert unterschritten, verlieren die Mikrotubuli ihre Integrität vollständig, was die betroffene Zelle zerstört. Dieses hyperphosphorylierte Tau-Protein kann nun (ähnlich wie ein Prion) in andere Zellen wandern und andere Tau-Proteine infizieren. Zudem kann es zu sogenannten NFTs (neurofibrilläres Bündel) in den Zellen verklumpen, was die Funktion der Zelle vor ihrem Tod noch weiter beeinträchtigt. Es scheint also einen Zusammenhang zwischen der Alzheimererkrankung und dem Auftreten von hyperphosphorylierten Tau-Proteinen im Gehirn zu geben und Amyloid-Plaques scheinen die Entwicklung dieser Tau-Proteine zu katalysieren. Aber das Immunsystem des Gehirns spielt hier nicht nur die Rolle des Schadensbegrenzers, denn im Moment gehen wir davon aus, dass die Mikroglia sogar dabei helfen, die Amyloid-Plaques zu verkleben und so die möglichen Schädigungen zu verstärken. Ob Alzheimer somit der „Kollateralschaden“ bei der Behandlung der Entzündungen ist, wird derzeit intensiv erforscht. Und hier kommen wir zur genetischen Komponente der Erkrankung.
Die Genetik
Menschen, die an Trisomie-21 (Down-Syndrom) leiden, haben 3 Kopien des 21. Chromosoms. Das Gen, das APP produziert, liegt auf Chromosom 21. Daher wird im Körper von Menschen mit Trisomie-21 mehr APP produziert. 30% der 50-jährigen Menschen mit Trisomie-21 leiden an Alzheimer. Bei den über 60-jährigen sind es bereits 50%. Damit liegen sie weit über dem Bevölkerungsdurchschnitt. Es drängt sich somit die Vermutung auf, dass die Amyloid-Plaques tatsächlich eine massive Rolle bei der Entwicklung von Alzheimer spielen. Grund genug, die genetische (und damit die familiäre) Komponente bei Alzheimer weiter zu untersuchen.
Ein weiteres Gen, das einen massiven Einfluss auf das individuelle Alzheimerrisiko hat, ist das Gen, das für das Apolipoprotein E (APOE) codiert. APOE spielt eine Rolle im Fettsäurestoffwechsel und kommt in der Bevölkerung in 3 Varianten vor. APOE ε2, APOE ε3 und APOE ε4. Und das hat signifikante Einflüsse auf Alzheimer:
Die ε4-Variante des Gens findet man in ungefähr 14% der Bevölkerung. Bei Alzheimerpatienten findet man diese Variante allerdings bei etwa 37% der Untersuchten. Im Gegensatz dazu findet man die ε2-Variante in 8% der Bevölkerung und nur 4% der Alzheimerpatienten. Die ε3-Variante findet man in der Bevölkerung am häufigsten, nämlich bei etwa 77% und bei etwa 60% der Alzheimerpatienten.
Es zeigt sich, dass für Menschen, die über die ε4-Variante des APOE-Gens verfügen, das Alzheimerrisiko also signifikant höher ist als für die, die eine der beiden anderen Genvarianten tragen. Und eine Ursache dafür könnten unsere eigenen Immunzellen sein. Denn insbesondere Mikroglia und Astrozyten (Stützzellen des Nervensystems) produzieren APOE, das dazu dient, Fette und Cholesterin in die Hirnzellen zu schleusen, die dort für die Bildung und Reparatur von Synapsen, sowie zur Stabilisierung ihrer Zellmembran genutzt werden. Über verschiedene Wege, zum Beispiel LDL-Rezeptoren wird APOE, das mit Cholesterin beladen ist, in die Zelle transportiert. Normalerweise würde APOE nun größtenteils abgebaut werden. In der ε4-Variante wird weniger Cholesterin von APOE aufgenommen, was den Zufluss ins Zellinnere reduziert. Das ist nur ein geringes Problem, da die Nervenzellen selbst ebenfalls Cholesterin produzieren können. Im Zellinneren neigt APOE ε4 allerdings dazu, zu verklumpen. Sein Abbau in den Lysosomen ist gestört; anstatt abgebaut zu werden, verklumpt das APOE dort im sauren Milieu sogar noch leichter, was neuroinflammatorische Signalkaskaden auslösen kann.
Aber auch außerhalb der Zelle spielt APOE eine Rolle. Hier werden APOE-Moleküle genutzt, um überschüssiges Cholesterin von den Nervenzellen weg und zu anderen Nervenzellen hinzutransportieren. In der ε4-Variante des Proteins geschieht allerdings seltsames. Denn durch die gestörte Affinität zu Cholesterin kann es dieses auch nicht mehr so schnell von der Zellmembran abtransportieren. Das überschüssige Cholesterin sammelt sich in der Zellmembran in sogenannten Lipid-Rafts an, die die Zellmembran durchdringen. Diese cholesterinreichen Regionen ziehen explizit die Enzyme β‑Sekretase und γ‑Sekretase an, da diese sehr lipophil sind. Lagert also APP zufällig in diesen Lipid-Rafts, steigt die Wahrscheinlichkeit, dass es fehlerhaft geschnitten wird. Die fehlerhaften Amyloid-β-Monomere werden nun in den Zwischenzellraum abgegeben, wo sie wiederum mit APOE zusammenlagern können. Eigentlich soll dieses APOE den Abbau der Amyloid-Plaques sogar unterstützen, da durch die Bildung des APOE-Amyloid-Komplexes ein Abbau durch Mikroglia oder ein Ausschleusen über die Blut-Hirn-Schranke ermöglicht wird. Nur haben wir hier ein weiteres Problem:
Der APOE-Amyloid-Komplex kann sich natürlich auch mit anderen Komplexen im Zwischenzellraum zusammenlagern, wodurch auch hier das Immunsystem aktiv wird und entzündliche Prozesse in Gang gesetzt werden. Neuronen können zudem nicht erkennen, ob APOE eine normale Cholesterinladung trägt, oder ob es mit Amyloid-β beladen ist. Also werden diese Komplexe auch versehentlich von Neuronen aufgenommen. Sie verklumpen im inneren der Neuronen, womit wir wieder beim Verhalten von ε4 im Zellinneren wären. Es scheint also, dass die Mikroglia selbst zur neurologischen Schädigung beitragen, indem sie unaufhörlich die fehlerhafte APOE-Variante produzieren. Gleichzeitig sollen Mikroglia und Astrozyten aber doch dabei helfen, die Amyloid-Plaques abzubauen.
Man kann sagen, dass die ε4-Variante von APOE das Alzheimerrisiko also drastisch erhöht. Angesichts dieser Daten gehen einige Menschen so weit zu sagen, dass die ε2-Variante von APOE aufgrund ihrer geringen Häufigkeit bei Alzheimerpatienten verglichen mit der Bevölkerung sogar eine Art Schutz vor Alzheimer bietet, während die ε3-Variante die Basis darstellt, die man sowohl in der Bevölkerung als auch den Patienten findet und die weder Schutz noch ein erhöhtes Risiko vor der Erkrankung bietet. In gewisser Hinsicht ist dies korrekt, da sowohl ε2 und ε3 auf normale Weise mit dem Rezeptor ABCA1 interagieren, der überschüssiges Cholesterin aus der Zelle schleust und ebenfalls die Amyloid-Plaques besser abbauen können.
Bei genauerem Hinsehen ist die Sache allerdings komplexer. So zeigen sich in Untersuchungen, dass z.B. Afrikaner mit der ε4-Variante trotzdem seltener an Alzheimer erkranken als Kaukasier oder Asiaten. Vermutungen zur Ursache dieser Unterschiede reichen von anderen genetischen Faktoren (die wir hier außen vor lassen), bis zur Ernährungsweise. Aber klar ist nur, dass wir weiterforschen müssen.
Wie gesagt, bisher handelt es sich bei all diesen Informationen um Arbeitshypothesen, bei denen noch kein endgültiger, kausaler Nachweis erbracht werden konnte. Wir schleichen uns lediglich auf Samtpfoten an die grundlegende Ursache heran. Aber warum wissen wir so wenig über Alzheimer? Der Namensgeber Alois Alzheimer beschrieb die Krankheit bereits 1901. Und über 120 Jahre später haben wir das menschliche Genom entschlüsselt, das Atom gespalten und scheitern noch immer daran, den Mechanismus einer Krankheit aufzuschlüsseln, an der jährlich fast 60.000 Menschen allein in Deutschland sterben.
Fehlschläge sind in der wissenschaftlichen Forschung und Medikamentenentwicklung ja an der Tagesordnung. Sie sind notwendig, ohne sie gäbe es keinen Fortschritt. Und manchmal entscheiden sich Forscher auch dazu, Holzwege zu Ende zu gehen. Zum Beispiel indem sie sich entscheiden, weiter an Medikamenten zu forschen, deren Nutzen nicht oder kaum vorhanden ist. All das ist absolut legitim und es ist mir wichtig, mich an dieser Stelle zu wiederholen.
Aber vielleicht ist es jetzt an der Zeit, über die Wissenschaftler zu reden, die bis vor Kurzem teilweise an der Spitze der Forschung zu dieser Krankheit standen und die im Verdacht stehen, versucht zu haben, ihre Holzwege als Autobahn zu verkaufen…
Alles nur Versehen
Beginnen wir mit Eliezer Masliah, dem Mann, der am National Institute on Aging (NIA) die sogenannte Division of Neuroscience geleitet hat. Und dabei auf einem Budget von $2,6 Milliarden (!) saß. Eine sehr einflussreiche Position, mit der er nicht nur die Forschungsschwerpunkte seines Instituts lenken konnte, sondern ebenfalls weltweite Impulse setzte.
Und eine erhebliche Zahl dieser Impulse basiert wohl auf gefälschten Daten. Denn ein nun erhobener Vorwurf lautet, dass Masliah oder ein Mitarbeiter seiner Forschungsgruppe in großem Stil die Ergebnisse von Western Blots und Mikroskopaufnahmen in über 130 Studien manipuliert haben soll. Ziel der Manipulation waren Daten des Proteins „Alpha-Synuclein“.
Alpha-Synuclein ist ein Protein, das in Synapsen vorkommt und die Freisetzung von Neurotransmittern – insbesondere Dopamin – mitreguliert. Veränderungen in Alpha-Synuclein werden mit Parkinson, Lewy-Körper-Demenz und anderen neurodegenerativen Erkrankungen in Verbindung gebracht.
Die Arbeit von Masliah und seinem Team war maßgeblich für die Prominenz dieser Hypothese verantwortlich und führte schließlich sogar zu einem Medikament, Prasinezumab, das noch immer in klinischen Studien an Patienten getestet wird, obwohl es schon in einer ersten großen Studie keinerlei Nutzen gezeigt hat.
Wer zu diesen Entwicklungen mehr Details möchte, sei auf diese Zusammenfassung der Situation verwiesen. Wir widmen uns nun dem nächsten Kandidaten.
Karen Ashe arbeitet an der University of Minnesota, wo ihr Fachgebiet die Alzheimerforschung ist. Im Jahr 2002 stellt sie einen Postdoc ein, der eine bahnbrechende Entdeckung machen wird. Ihr Protegé Sylvain Lesné findet 2006 im Hirngewebe von Mäusen das Protein Aβ*56. Ein schweres Amyloid-Stück, das bei Mäusen, die eine große Zahl davon im Gehirn haben, dafür sorgt, dass sie keine neu erlernten Informationen abrufen können. Bahnbrechend! Ein Durchbruch! Da liegt ein Nobelpreis in der Luft! Dieses Ergebnis hat vor ihm noch nie jemand zustande gebracht.
Naja, genaugenommen hat es wohl bis heute niemand zustande gebracht. Denn kein Labor konnte Lesnés Ergebnisse reproduzieren. Trotzdem war der initiale Hype um das Ergebnis massiv. Als Laborleiterin gewann Ashe 2006 den mit $100.000 dotierten Potamkin-Preis, ihr Labor bekam eine Spende in Höhe von $5 Millionen, gestiftet von Beverly Grossman, der Witwe des an Alzheimer verstorbenen Anteilseigners an den Minnesota Vikings, Bud Grossman.
Diese Euphorie war auch nicht unberechtigt. Dass Amyloid-Plaques irgendwas mit der Erkrankung zu tun haben, sollte in diesem Artikel bisher klar geworden sein. Jetzt genau das Partikel zu finden, das als vielversprechender Auslöser galt, war unter dieser Prämisse also nur eine Frage der Zeit.
Mittlerweile hat sich herausgestellt, dass es ebenfalls nur eine Frage der Zeit war, bis die manipulierten Bilder in der Arbeit von Lesné und Ashe auffliegen würden. Auch wenn der Urheber der Manipulationen nicht nachgewiesen werden konnte.
2024 kamen die Vorwürfe ans Licht, die bahnbrechende Studie wurde zurückgezogen und bis zuletzt hat sich Lesné (anders als die anderen Autoren) gegen eine Rücknahme der Veröffentlichung gestemmt. Erfolglos. Der Mann, dem das NIH in Minnesota ein Labor finanzierte, wurde anschließend nach einer mehrjährigen Untersuchung vorübergehend beurlaubt. Lesné hat seit dem 1. Mai 2025 keine wissenschaftliche Position an der University of Minnesota mehr inne. Denn es gerieten noch knapp 20 weitere Arbeiten von Lesné unter Verdacht, von denen aber nur ein Teil gemeinsam mit Ashe entstanden ist. Aber keine Arbeit war so prominent, wie die, die er 2006 veröffentlicht hat.
Die Bedeutung dieser Arbeit kann dabei nicht überbewertet werden. Seit der Veröffentlichung dieser Studie hat das NIH bis 2021 fast $300 Millionen für die Erforschung dieser Plaques zur Verfügung gestellt. Das Geld ging dabei nicht an Lesné allein, sondern an eine Vielzahl von Projekten, die Plaques erforscht haben. Die Forschungsergebnisse von Lesné und seinen Kollegen waren aber wegweisend, die Plaques ins Zentrum der Alzheimerforschung zu stellen. Sie waren nicht allein dafür verantwortlich, aber sie haben dem Feld den Anschub gegeben, den es brauchte.
Verantwortlich dafür, dass dieser Betrug, von dem bis heute nicht klar ist, wer die Hauptschuld trägt, überhaupt aufgeflogen ist, ist, wie auch schon bei Cassava, der Wissenschaftler Matthew Schrag, der seine Karriere aufs Spiel gesetzt hat, um diesen Betrug zu veröffentlichen.
Es ist ebenfalls unmöglich über Matthew Schrag zu sprechen, ohne ebenfalls Elisabeth Bik zu erwähnen. Sie ist ein wichtiger Teil einer kleinen, aber lautstarken Gruppe, die in der biologischen Forschung viele Arbeiten auf „Unachtsamkeiten“ untersucht und die ein oder andere wissenschaftliche Karriere beendet hat. Auch wenn dies mit entsprechendem Gegenwind aus der Forschergemeinde einhergeht. Es gibt kaum einen Artikel, der einen Namen ohne den anderen erwähnt.
Ein weiterer Forscher, der sein Labor wohl nicht ordentlich im Griff hatte, ist Marc Tessier-Lavigne, der ehemalige Präsident der Stanford-University und ehemaliger Chief Scientific Officer bei Genentech. 2022 veröffentlichte der Nachwuchsjournalist Theo Baker eine Reihe von Artikeln, die zeigten, dass Tessier-Lavigne Co-Autor mehrerer neurobiologischer Studien war, in denen gezielt Bilder manipuliert wurden. Der Urheber der Manipulationen konnte natürlich nie festgestellt werden. Beteiligt an der Aufdeckung dieser Manipulationen war auch hier wieder Elisabeth Bik.
Infolgedessen wurden einige Paper zurückgezogen, unter anderem auch eines aus dem Februar 2009. Dieses Paper war maßgeblich dafür verantwortlich, dass Genentech ein eigenes Alzheimermedikament erforscht hat. Dessen Entwicklung wurde 2012 eingestellt, weil die grundlegende Idee, die sich mittlerweile als Manipulation herausgestellt hat, nicht verlässlich reproduziert werden konnte.
Die Vorwürfe an Tessier-Lavigne lauten hierbei explizit nicht, dass er selbst Daten manipuliert hätte, aber dass er nicht genug getan hat, als er auf diese Manipulationen mehrfach aufmerksam gemacht wurde.
Für weitere Details möchte ich auch hier wieder auf einen weiterführenden Artikel verweisen.
Ebenfalls unter Verdacht steht Berislav Zloković, Professor für Neurowissenschaften an der Keck School of Medicine und Mitbegründer des Unternehmens ZZ Biotech, das ein experimentelles Schlaganfallmedikament entwickelt. Zudem hat Zloković ebenfalls an Alzheimer geforscht.
2023 wandten sich Whistleblower mit einem 113-Seitigen Dossier an die Behörden, in dem sie darlegen, wie Zloković in seinem Labor explizit Mitarbeiter angewiesen haben soll, Messwerte anzupassen und Bilder zu manipulieren, um die Ergebnisse zu zeigen, die Zloković genehm waren. Die Vorwürfe sind nicht belegt. Betroffen sind viele Paper, die über eine Spanne von mehr als 25 Jahren veröffentlicht wurden. Dies soll sich ebenfalls auf die Daten des Schlaganfallmedikaments erstreckt haben, weshalb die US-Behörden die Zulassungsstudien aussetzen und nun ihrerseits eine Untersuchung durchführen, um die Vorwürfe zu prüfen. Patienten, die das Medikament in einer Phase-II-Studie bekamen, waren nach der 90-tägigen Versuchsdauer eher auf äußere Hilfe angewiesen, als die Placebogruppe, zudem starben in der ersten Woche nach Gabe des Medikaments 6 von 66 Menschen in der Medikamentengruppe, aber nur 1 von 44 in der Placebogruppe.
Zurückgezogen wurden auch einige seiner Arbeiten über Alzheimer, wo er sich z.B. damit befasste, wie die Blut-Hirn-Schranke angeblich beim Abtransport von Amyloid-Plaques helfen sollte. Angesichts der Vorwürfe bezüglich des Schlaganfallmedikaments stehen die Vorwürfe bezüglich seiner Alzheimerforschung selbstverständlich hinten an.
Die bisherigen Beispiele sind längst nicht abschließend, zeigen aber, wie eine Gruppe von Forschern, egal wer sie genau sein mögen, die Forschung an der Alzheimer-Krankheit in eine bestimmte Richtung gelenkt und so massive Schäden angerichtet hat. Egal ob wir diesen Betrug von Seiten der Patienten, Angehörigen, Forscher, die keine Fördermittel bekamen, öffentlichen Geldgeber oder privaten Unternehmen betrachten, sie alle wurden betrogen. Ein Betrug, der noch Jahrzehnte nachhallen wird, während die Forschung sich endlich wieder in andere Gebiete ausbreitet und das Vertrauen in die Alzheimerforschung langsam wiederhergestellt werden muss.
Auch wenn klar sein sollte, dass in der Amyloid-Hypothese ein wahrer Kern stecken muss, wäre diese Hypothese ohne den vielfachen Betrug nie so prominent geworden. Wir könnten also heute vielleicht schon Medikamente haben, die besser wirken als die, die derzeit zugelassen sind.
Neue Medikamente
Eines der wenigen Medikamente, die in der Öffentlichkeit als vielversprechend gelten und auch in Deutschland zugelassen ist, ist Lecanemab. Der Antikörper soll die Anhäufung der Amyloid-Plaques im Gehirn verlangsamen.
Um zu vergleichen, ob das Medikament den kognitiven Abbau verlangsamt, wurde in der Zulassungsstudie ein Alzheimer-Scoring verwendet, der in 6 Kategorien (u.a. Gedächtnis, Orientierung, persönliche Pflege) jeweils 0-3 Punkte zuordnet. Je höher die Punktzahl, desto stärker der kognitive Abbau. Die Gruppe, die Lecanemab erhalten hat, hat in den 18 Monaten Studiendauer im Schnitt 1,21 Punkte erreicht, die Placebogruppe 1,66. Die Zulassung des Medikaments beruht also auf der Differenz dieser Scores, von 0,45 Punkten, was eine relative Verzögerung des Krankheitsverlaufs von etwa 27% bedeutet. Ob dies im Alltag der Patienten einen Unterschied macht, ist bis heue fraglich.
In der Medikamentengruppe kam es zu 6 Todesfällen, in der Placebogruppe zu 7. Auch wenn die Zahlen ähnlich sind, gab es mehrere Veröffentlichungen zu den Todesfällen in der Medikamentengruppe. So hat man festgestellt, dass Träger von 2 Kopien des APOE-ε4-Gens ein erhöhtes Risiko für Blutungen im Gehirn und Hirnschwellungen haben, weshalb vor Einsatz des Medikaments ein Gentest für jeden Patienten verpflichtend ist, denn wer 2 Kopien des APOE-ε4-Gens und damit das höchste Alzheimerrisiko hat, darf das Medikament nicht bekommen.
Insgesamt kam es in der Studie in rund 13% der Fälle zu Hirnschwellungen, in etwa 17% der Fälle kam es zu Blutungen im Gehirn, weshalb die Gabe des Medikaments nur verbunden mit regelmäßigen MRT-Untersuchen erfolgen darf, die solche Veränderungen vorab feststellen sollen.
Zudem fand man in der Medikamentengruppe eine Reduzierung des Hirnvolumens, die größer war als bei Alzheimerpatienten der Placebogruppe.
Da das Medikament auch in Deutschland zugelassen ist, sind diese Informationen für Patienten relevant, um eine Abwägung bezüglich der Medikation treffen zu können.
Zwei weitere Antikörper, Aducanumab und Donanemab sind in Deutschland nicht zugelassen, aber in den USA. Die Erläuterung der beiden Medikamente wäre quasi identisch zum Absatz über Lecanemab, nur müssten wir die Zahl der Patienten mit Nebenwirkungen hochschrauben. Aufgrund der fehlenden Zulassung in Deutschland sparen wir uns das aber.
Die Früherkennung
Bei dem durchwachsenen Ausblick auf Forschungsergebnisse und Behandlungsmethoden bleibt ja nur der Blick auf die möglichst frühzeitige Vermeidung von Alzheimer.
Eine Möglichkeit sind Frühtests, die darauf abzielen, die Biomarker für Alzheimer rechtzeitig zu erkennen und dann durch Therapien gegensteuern zu können. Und weil mir dieser Abschnitt des Artikels besonders am Herzen liegt, möchte ich gleich das Fazit vorwegnehmen:
Wir haben keine Biomarker, die eindeutig belegen, dass ein Patient tatsächlich an Alzheimer leidet. Ein alleiniger, positiver Befund durch Biomarker ist nicht ausreichend, um eine konkrete Erkrankung festzustellen, geschweige denn eine Therapie zu rechtfertigen. Schon gar keine medikamentöse Therapie! Wir können maximal sagen, dass das Vorhandensein von Biomarkern ein erhöhtes Risiko darstellt, aber wer keinen kognitiven Abbau zeigt, leidet nicht an Alzheimer. Da diese Diagnose aber mit einer massiven Einbuße der Lebensqualität einhergeht, wird sie die Menschen in Therapien treiben, egal ob zugelassen oder nicht, egal ob echte Medikamente oder Präparate von Scharlatanen. Und jetzt packen wir das in Zahlen:
Vor kurzem haben britische Forscher beispielsweise mit einem MRT-Modell von sich reden gemacht, das angeblich bereits 9 Jahre vor Ausbruch der Krankheit erste Veränderungen im Gehirn erkennen kann. Und zwar mit einer Genauigkeit von bis zu 84%. Und das bis zu 9 Jahre im Voraus! Klingt das nicht toll?
Aber was das in der Realität bedeutet, lässt sich mit dem Satz von Bayes ausdrücken. Einer Methode aus der Statistik, mit der wir untersuchen, ob ein hohes Level an positiven Befunden auch ein hohes Level an Erkrankten bedeutet. Wir erläutern das Prinzip zunächst an einer vereinfachten Überschlagsrechnung, in den darauffolgenden Tabellen wurde der Satz aber korrekt angewendet.
Tun wir mal so, als würden von 100 Leuten 10 an Demenz erkranken. Die Inzidenz läge also bei 10%. Der Test erkennt 84% der Erkrankten korrekt und 16% nicht korrekt. Auf unsere Gruppe von 100 Menschen bezogen, bei denen 10 an Demenz leiden und 90 gesund sind, bedeutet das folgendes: Der Test erkennt also etwa 8 Leute, die tatsächlich Alzheimer haben (8 von 10 Erkrankten) und stellt bei weiteren 16 Leuten eine falsch-positive Diagnose. Insgesamt haben also 24 Menschen eine Demenz-Diagnose, von denen aber nur 8 Leute tatsächlich erkranken werden. Wer also eine positive Diagnose durch diesen Test bekommt, hat lediglich eine Wahrscheinlichkeit von etwa 30%, auch tatsächlich an Demenz erkrankt zu sein. Nicht gerade ein vertrauenserweckender Durchbruch. Denn die Inzidenz von Demenz streut natürlich innerhalb der Bevölkerung recht breit. Da die Diagnostik aber bis zu 9 Jahre vor Ausbruch der Krankheit erfolgen soll, lässt sich davon ausgehen, dass dieser Test in erster Linie von Menschen wahrgenommen wird, die noch unter 85 sind und damit nicht in die Hochrisikogruppe für Demenz fallen, was bedeutet, dass die reale Inzidenz nicht bei 10% liegt, die ich für die Rechnung angenommen habe, sondern deutlich darunter.
Aber wie gesagt: Die Alzheimerinzidenz streut breit. Während etwa 33% der über 85-jährigen an Alzheimer leiden, sind es bei den über 70-jährigen nur etwa 4%. Noch geringer ist die Inzidenz bei den unter 60-jährigen, wo sie bei etwa 1% liegt. In der Allgemeinbevölkerung sind es insgesamt etwa 5-8% der Menschen. Die Qualität der Früherkennung hängt also stark vom Alter ab. Wir berechnen nun die Daten für diverse Testmethoden. Wir nehmen dabei an, dass in der allgemeinen Bevölkerung sogar 10% der Leute erkrankt seien. Diese Zahl ist immens überhöht, aber dann kann uns niemand vorwerfen, die Testmethoden schlechtzurechnen.
Vergleichen wir die Zahlen für das britische MRT, die wir eben überschlagen haben. Ich runde die Wahrscheinlichkeiten dabei immer auf, bzw. ab. Für die, die nachrechnen wollen, verwendet habe ich folgende Werte: Sensitivität 84%, Spezifität 90% (steht nicht in der Studie, ist einfach nur eine wohlwollende Schätzung).
Tab.1: fMRT Alzheimerprävalenz und Testwahrscheinlichkeit
| Alter | Prävalenz | Wahrscheinlichkeit auf positiven Test | Wahrscheinlichkeit, dass das Ergebnis falsch-positiv ist |
| Allgemeinbevölkerung | 10% | 17% | 52% |
| Menschen unter 60 | 1% | 11% | 93% |
| Menschen über 70 | 4% | 13% | 76% |
| Menschen über 85 | 33% | 35% | 19% |
Ohne zusätzliche Risikofaktoren, z.B. den kognitiven Abbau, genetische Risikofaktoren oder Vorerkrankungen können wir also sagen, dass der Test mit einer ernstzunehmenden Wahrscheinlichkeit in jeder dieser Gruppen ein positives Ergebnis zeigt (Spalte 3), aber in der Realität die Wahrscheinlichkeit dafür, dass es sich um ein falsch-positives Ergebnis handelt, immens höher ist (Spalte 4). Die einzige Ausnahme ist die Hochrisikogruppe für Menschen über 85, da hier die Prävalenz von Alzheimer sehr hoch ist und die Tests hier besser abschneiden.
Wir betrachten als nächstes Plasma phospho-tau217, ein Biomarker, der seit Jahren in der Diagnostik verwendet wird. Hier basieren die Daten zur Berechnung auf einem wohlwollenden Durchschnitt diverser Validierungen: Sensitivität 91%, Spezifität 94%
Tab. 2.: p-tau217 Alzheimerprävalenz und Testwahrscheinlichkeit
| Alter | Prävalenz | Wahrscheinlichkeit auf positiven Test | Wahrscheinlichkeit, dass das Ergebnis falsch-positiv ist |
| Allgemeinbevölkerung | 10% | 15% | 37% |
| Menschen unter 60 | 1% | 7% | 87% |
| Menschen über 70 | 4% | 9% | 61% |
| Menschen über 85 | 33% | 34% | 11% |
Wir sehen eine Verbesserung der Zahlen im Vergleich zur MRT-Methode, aber auch hier ist die Wahrscheinlichkeit auf ein falsch-positives Ergebnis in den Gruppen mit dem geringsten Risiko sehr hoch. Und als nächstes schnappen wir uns CSF Aβ₄₂, ebenfalls ein beliebter Biomarker. Wir nehmen wieder Durchschnittsdaten zum Vorteil der Testmethode: Sensitivität: 80%, Spezifität 82%
Tab. 3.: CSF Aβ₄₂ Alzheimerprävalenz und Testwahrscheinlichkeit
| Alter | Prävalenz | Wahrscheinlichkeit auf positiven Test | Wahrscheinlichkeit, dass das Ergebnis falsch-positiv ist |
| Allgemeinbevölkerung | 10% | 24% | 67% |
| Menschen unter 60 | 1% | 19% | 96% |
| Menschen über 70 | 4% | 20% | 84% |
| Menschen über 85 | 33% | 38% | 31% |
Wir könnten noch weitere Biomarker testen, aber das Bild ist klar. Die Wahrscheinlichkeit, dass ein Biomarker Alzheimer vorhersagt, ist für Leute unter 60 extrem gering. Erst mit über 80 steigt die Aussagekraft auf ein halbwegs vernünftiges Niveau an. Wer also gesund ist, unter 60, keine genetischen Prädispositionen (z.B. ε4) hat und auch in der Familie keine Alzheimerfälle vorweisen kann, ist gut beraten, sich vorher genau zu überlegen, ob er so einen Test machen will und was dessen Aussagekraft ist. Insbesondere im Hinblick auf die bisherigen Therapiemethoden. Wir könnten uns noch andere Tests anschauen, z.B. den CDR-SB-Score, der auch für die Zulassung von Lecanemab verwendet wurde. Der soll bis auf 8 Monate genau vorhersagen, ob jemand Alzheimer entwickelt. Aber auch hier braucht es noch mehr Validierung und vor allem mehr Replikationen der Ergebnisse.
Jenseits des Amyloids
Hört man sich unter Alzheimerforschern um, so entsteht der Eindruck, dass ein großer Teil dieser Wissenschaftler der Ansicht ist, dass der Amyloid-Hypothese zu viel Raum eingeräumt wurde, obwohl nicht klar ist, wie groß der Anteil des Amyloids an der Entstehung von Alzheimer tatsächlich ist.
Wir haben in den letzten Abschnitten viele überzeugende Hinweise gesehen, die zeigen, dass Amyloid-Plaques definitiv eine Rolle bei der Entstehung und Entwicklung von Alzheimer spielen müssen. Aber wäre dies der Weisheit letzter Schluss, müssten Medikamente, die diese Plaques angreifen, eine Wirkung zeigen. Und die sehen wir nicht. Zumindest nicht in dem Maße, in dem wir sie erwarten würden.
Also möchte ich diesen Abschnitt der Spekulation widmen, wohlwissend, dass vieles von dem, das ich hier auflisten werde, in den nächsten Jahren im Sande verlaufen wird. Aber wer weiß, vielleicht erweist sich ja eines der folgenden Themen als Schlüssel, der zu einer wirksamen Behandlung oder sogar Heilung führen wird. Dann kann ich sagen: Ich hab’s euch ja gesagt!
Die cholinerge Hypothese
Eine der frühesten Entdeckungen im Zusammenhang mit neuronaler Degeneration war die der cholinergen Neuronen. Diese Neuronen finden sich im Nucleus basalis Meynert, einer Hirnregion, die für das bewusste Lernen zuständig ist. Hier schütten diese Neuronen den Neurotransmitter Acetylcholin aus, der für die Gedächtnisleistung relevant ist. Eine Degeneration dieser cholinergen Neuronen führt also zu einem Nachlassen der kognitiven Leistungsfähigkeit.
Um Acetylcholin zu synthetisieren, sind die Neuronen auf ein Enzym namens ChAT angewiesen, das bei Alzheimerpatienten stark reduziert ist. Dabei korreliert die Reduktion des Enzyms mit dem kognitiven Abbau.
Könnten wir also die Konzentration an Acetylcholin im Gehirn erhalten, könnte dies den neuronalen Abbau wenigstens temporär verlangsamen. Deshalb steht an vorderster Front der Alzheimer-Medikation seit Jahren die Gruppe der Acetylcholinesterasehemmer, die genau dies tun. Zumindest temporär, für 12-18 Monate. Sie verringern die Abbaugeschwindigkeit des Acetylcholins, können aber weder die Menge an produziertem Acetylcholin erhöhen, noch die zugrundeliegende Krankheit aufhalten. Da Acetylcholin im Körper aber noch einige andere Aufgaben erfüllt, ist zudem die mögliche Dosierung des Medikaments stark begrenzt. Gerade weil das Nebenwirkungsprofil der Acetylcholinesterasehemmer gut bekannt ist, sind sie noch heute eines der ersten Medikamente, die zur Verzögerung der Erkrankung verschrieben werden.
Eine Frage, welche die Forschung aber nicht beantworten konnte, ist das Henne-Ei-Problem. Was kommt zuerst? Lassen die Neuronen nach, was zu Alzheimer führt, oder führt Alzheimer zu einem Nachlassen der Neuronen?
Zugegeben, die Frage ist auch heute noch Gegenstand der Forschung, aber Acetylcholin erlebt ein kleines Revival als mögliches Medikament gegen das Voranschreiten von Alzheimer, mit neuen Präparaten, die die Nebenwirkungen reduzieren und so vielleicht das Interesse der Forscher neu entfachen können.
Die Mitochondrienkaskade
Eine der Ideen, die in den letzten Jahren an Zulauf gewonnen haben, ist die Mitochondrienkaskade. Die Idee geht davon aus, dass die Genetik eines Menschen die Aktivität und Resilienz der Mitochondrien bestimmt. Bei einigen sind diese Faktoren stärker ausgeprägt, bei anderen nicht so stark. Da die Mitochondrienfunktion auf natürliche Weise mit dem Alter abnimmt, führt dies dazu, dass die Mitochondrien einiger Menschen schneller altern. Irgendwann ergibt sich für einen Menschen also ein funktionelles Mindestniveau, das sie nicht unterschreiten können, ohne Krankheitssymptome zu zeigen, die wir mit Demenz bzw. speziell mit Alzheimer verknüpfen. Bei einigen Menschen passiert dies früher, bei anderen später. Diese Aussage basiert auf vielen Forschungsergebnissen, die die nachlassende Aktivität der Mitochondrien in einen Zusammenhang mit der Tau-Phosphorylierung und dem Auftreten von Amyloid-Plaques bringen. Die Neurodegeneration steht also in einem direkten Zusammenhang zum Abbau der Mitochondrien, bedingt durch die Genetik und natürlich Umweltfaktoren. Bisher haben wir hierfür noch keine überzeugenden Daten im Menschen, lediglich Tiermodelle und Ergebnisse, die als Nebenprodukt von anderen Studien abgefallen sind. Die wichtige Frage, die es zu beantworten gilt, ist, ob die Mitochondrien abbauen und so zur Alzheimerdemenz führen, oder ob die Alzheimerdemenz den Mitochondrienabbau verstärkt.
So oder so, mehr Forschung in diese Richtung wird vielleicht nicht zur Heilung der Krankheit führen, aber definitiv für ein besseres Verständnis sorgen, und wenn das Verständnis auch nur darin besteht, ein weiteres Modell ausschließen zu können.
Herpes-Simplex-Viren (HSV)
Jap. Korrekt. Herpes. Die Idee, dass eine latente Infektion mit Herpes-Viren eine Rolle bei der Entwicklung von Alzheimer spielt, ist nicht neu. Bereits vor rund 30 Jahren hat man festgestellt, dass HSV das Gehirn befallen kann. Schon 2018 hat man zudem in Taiwan eine Kohorte von 33.000 Menschen auf den Zusammenhang zwischen Herpes und Alzheimer untersucht und stellte fest, dass diejenigen, bei denen eine Herpes-Infektion vorlag, ein 2,5-mal so großes Risiko einer Alzheimererkrankung hatten, wie die herpesfreie Kontrollgruppe. Interessant ist, dass dieses Risiko um 90% reduziert werden konnte, wenn die Patienten antivirale Medikamente erhalten haben.
Diese Ergebnisse werden von diversen Kohortenstudien (u.a. aus Südkorea, Schweden), sowie einer Metaanalyse basierend auf diversen Fallkontrollstudien unterstützt.
Besonders interessant ist hierbei, dass eine Kombinierte Infektion von HSV-1 mit dem Varizella-Zoster-Virus (VZV, dem Auslöser der Windpocken und der Gürtelrose, der ebenfalls Teil der HSV-Familie ist) das Risiko nochmal deutlich erhöht, was gerade im Hinblick auf die große Zahl derer, die mit VZV infiziert sind, zukünftige Forschungsimpulse setzen könnte.
Die derzeitige Hypothese, wie diese Viren Alzheimer auslösen, bzw. begünstigen lautet wie folgt:
Nach einer Infektion mit Herpes schlummern die Viren in den Ganglionen des Trigeminusnerven, der die Reizleitung der Gesichtsnerven, sowie die entsprechende Gesichtsmotorik steuert. Bei erneuter Aktivierung können die Viren dann über diesen Nerv ins Gehirn vordringen, wo dieses zum Schutz Aβ-Plaques bildet, die an die Oberflächenproteine der Viren binden und diese so deaktivieren. Zudem kann HSV-1 die Integrität der Blut-Hirn-Schranke beeinträchtigen, was das Eindringen weiterer Herpes-Viren erleichtert. Dabei zeigt sich auch, dass ein wesentlicher Faktor für die Schädlichkeit nicht nur die wiederholte Reinfektion mit HSV-1 ist, sondern die Genetik eine Rolle spielt. Patienten mit 2 Allelen von APOE-ε4 hatten erneut das größte Risiko für das Auftreten von Alzheimer in Zusammenhang mit einer Infektion. Ein Zusammenhang, der inzwischen mehrfach bestätigt wurde.
Ein wesentlicher Teil dieser Forschung geht auf Ruth Itzhaki zurück, die in einem Artikel bei STAT auch darüber spricht, wie man in Fragen der Forschungsförderung ihre Hypothese zugunsten der Amyloid-Plaques hintenangestellt hat.
An der Stelle möchte ich darauf hinweisen, dass es sehr leicht ist, diese These zu bestätigen, besonders weil es mittlerweile sehr viele Forschungsdaten zu den biochemischen Interaktionen zwischen dem HSV-Genom und den Hirnzellen gibt (insbesondere im Hinblick auf die Schädigung der Mitochondrien, die ja ebenfalls schon besprochen wurden). Trotzdem sollten wir nicht vergessen, dass wir bisher lediglich eine Synergie zwischen HSV und Alzheimer sehen, aber trotzdem mehr Forschung nötig ist.
TREM2
Wenn wir so über Mikroglia und ihre Rolle bei der Produktion von APOE sprechen, könnte man diese Immunzellen ja beinahe für den Feind halten. Dabei kann eine Aktivierung von Mikroglia gerade in frühen Stadien viele der negativen Effekte abpuffern, die durch APOE-ε4 entstehen. Hierfür bietet sich der Rezeptor TREM2 an, der auf der Oberfläche der Mikroglia zu finden ist und diese aktiviert. Die Idee ist, dass besonders in frühen Stadien von Alzheimer die Mikroglia noch in der Lage sind, die Mischung aus APOE und Aβ-Monomeren zu zerstören, um so das Voranschreiten von Alzheimer zu verlangsamen.
Die Idee wird intensiv erforscht, besonders weil bekannt ist, dass Mutationen in TREM2 auch das Alzheimerrisiko massiv erhöhen können. Erste Rückschläge hat die Idee allerdings auch schon erlitten. In einer klinischen Studie hat AbbVie Ende 2024 erfolglos ein Medikament getestet, das TREM2 aktivieren und so das Voranschreiten von Alzheimer bremsen sollte. Es hat die nötigen Endpunkte verfehlt, also keinen Vorteil gegenüber der Placebogruppe gezeigt.
Und wenn es doch die Plaques sind?
Jap. Was, wenn der ganze Stress umsonst war und es trotz allem die Amyloid-Plaques sind? Es gibt da ein besonderes Amyloid. Unser alter Bekannter Aβ42. Neuere Forschungsarbeiten deuten nämlich in die Richtung, dass dieses Amyloid die Wirksamkeit der γ-Sekretase reduziert. Was erstmal gut klingt, weil dadurch die Zahl der falsch geschnittenen Plaques abnehmen soll, ist ein Problem. Denn der Körper verwendet Enzyme gerne in verschiedenen Rollen. γ-Sekretase ist an vielen anderen Substraten beteiligt, sodass eine Inaktivierung schwere Folgen für die kognitive Funktion haben kann. Das wurde in einem geringeren Maße bereits durch Forschung von Eli Lilly gezeigt. Die haben einen γ-Sekretase-Inhibitor zur Behandlung von Alzheimer erforscht, aber in Phase III die Forschung abgebrochen, nachdem sich herausstelle, dass in der Medikamentengruppe die kognitiven Leistungen schneller abnahmen, als in der Placebogruppe. Es besteht also das Risiko, dass eine Inhibition der γ-Sekretase einer Kaskade gleicht, die andere Stoffwechselwege hemmt und so noch mehr toxische Produkte im Gehirn anreichert.
Fazit
Es bleibt das Wissen, dass Alzheimer uns noch lange beschäftigen wird. Der massive Forschungsbetrug hat uns vermutlich um mindestens 10 Jahre zurückgeworfen, auch wenn ein Teil dieses Betrugs darauf zurückzuführen ist, dass die Amyloid-Hypothese tatsächlich der vielversprechendste Ansatz ist. Immerhin werden nun auch Alternativen erforscht, die vielleicht ihren Teil zur Lösung des Rätsels beitragen. Wir haben noch immer keine wirklich langfristig wirksamen Medikamente, und die leisen Hoffnungsschimmer in Form moderner Medikamente müssen sich angesichts der Nebenwirkungen die Frage stellen, ob der Nutzen die Kosten rechtfertigt. So oder so, was bei der Erforschung von Alzheimer passiert ist, sollte uns alle aufwecken. Wir müssen noch genauer hinschauen, wo Forschung in großem Stil manipuliert wird. Das sind wir ehrlichen Forschern schuldig. Aber auch Patienten, die für diese Manipulation mit dem Leben bezahlen.